从SRA数据库下载原始数据
date: 2023-11-14 update slug: SRA_download_1 key: SRA,ENA,NCBI,EBI,prefetch,fasterq-dump cover: https://raw.githubusercontent.com/hermanzhaozzzz/PicturesBed01/master/PicGo/20231112193844.png ref: https://blog.csdn.net/yearstime/article/details/123719020
基本概念
SRA数据库
Sequence Read Archive:隶属NCBI (National Center for Biotechnology Information),它是一个保存大规模平行测序原始数据以及比对信息和元数据 (metadata) 的数据库,所有已发表的文献中高通量测序数据基本都上传至此,方便其他研究者下载及再研究。其中的数据则是通过压缩后以.sra文件格式来保存的,SRA数据库可以用于搜索和展示SRA项目数据,包括SRA主页和 Entrez system,由 NCBI 负责维护。
ENA数据库
European Nucleotide Archive:隶属EBI (European Bioinformatics Institute),功能等同SRA,并且对保存的数据做了注释,界面相对于SRA更友好,对于有数据需求的研究人员来说,ENA数据库最诱人的点应该是可以直接下载fastq (.gz)文件,由 EBI 负责维护。
SRA和ENA数据库是否互通?
两者在主要功能方面非常类似,同时数据互通。高通量数据分析时,需要从公共数据库如 NCBI、EBI 下载他人提交的高通量测序数据。
SRA数据库的名词概念
SRA 与 Trace 最大的区别是将实验数据与 metadata(元数据)分离。metadata 是指与测序实验及其实验样品相关的数据,如实验目的、实验设计、测序平台、样本数据(物种,菌株,个体表型等)。metadata可以分为以下几类:
Study【Project】
accession number 以 DRP,SRP,ERP 开头, 表示的是一个特定目的的研究课题,可以包含多个研究机构和研究类型等。study 包含了项目的所有 metadata,并有一个 NCBI 和 EBI 共同承认的项目编号(universal project id)【以PRJ开头!!】,一个 study 可以包含多个实验(experiment)。
Submission
一个 study 的数据,可以分多次递交至 SRA 数据库。比如在一个项目启动前期,就可以把 study,experiment 的数据递交上去,随着项目的进展,逐批递交 run 数据。study 等同于项目,submission 等同于批次的概念。
Sample
accession number 以 DRS,SRS,ERS 开头,表示的是样品信息。样本信息可以包括物种信息、菌株(品系) 信息、家系信息、表型数据、临床数据,组织类型等。可以通过Trace来查询。
Experiment
accession number 以 DRX,SRX,ERX 开头, 表示一个实验记载的实验设计(Design),实验平台(Platform)和结果处理(processing)三部分信息。实验是 SRA 数据库的最基本单元,一个实验信息可以同时包含多个结果集(run)。
Run
accession number 以 DRR,SRR,ERR 开头, 一个 Run 包括测序序列及质量数据。
下载公共数据集的原始数据示例
获取 Sample (GSM) / Series (GSE) / Project (PRJ) 编号
一般使用GEO或者PRJ等关键词检索文献,即可找到:


下面以PRJNA857874为例
安装使用sra-tools和entrez-direct工具集
conda install -c bioconda sra-tools entrez-direct
解析所有文件
mkdir fastq # 接下来去查询这个数据,解析到csv文件中 esearch -db sra -query PRJNA857874 | efetch -format runinfo > info.csv # 可以cat查看或者excel查看
下载SRA文件
其实cat可以知道,这个info.csv表里就一个文件,如果有多个文件可以进行循环下载,不再赘述
# cat查看可看到SRR20083768,这本质上是一个run # 后续跟了SRP385993,PRJNA857874,等编号 # 说明它同时属于这个SRP (STUDY Accession = SRP385993) # 和这个Project (BioProject Accession = PRJNA857874) # 使用prefetch下载这个run文件(SRR20083768),即可得到一个sra文件 mkdir sra && cd sra prefetch -X 100G -f no -p -o SRR20083768.sra SRR20083768 # -X|--max-size <size> # Maximum file size to download in KB (exclusive). Default: 20G # -f|--force <yes|no|all|ALL> # Force object download: one of: no, yes, all, ALL. # no [default]: skip download if the object if found and complete; # yes: download it even if it is found and is complete; # all: ignore lock files (stale locks or it is being downloaded by another process use at your own risk!); # ALL: ignore lock files, restart download from beginning. # -p|--progress # Show progress. # -o|--output-file <FILE> # Write file to FILE when downloading single file.
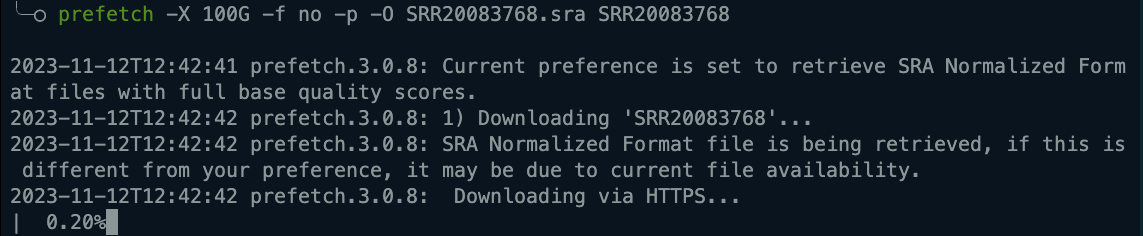
断点续传
注意了,prefetch自动支持断点续传,可以在正常下载途中,ctrl + c打断程序运行,然后重新执行相同的下载命令尝试一下
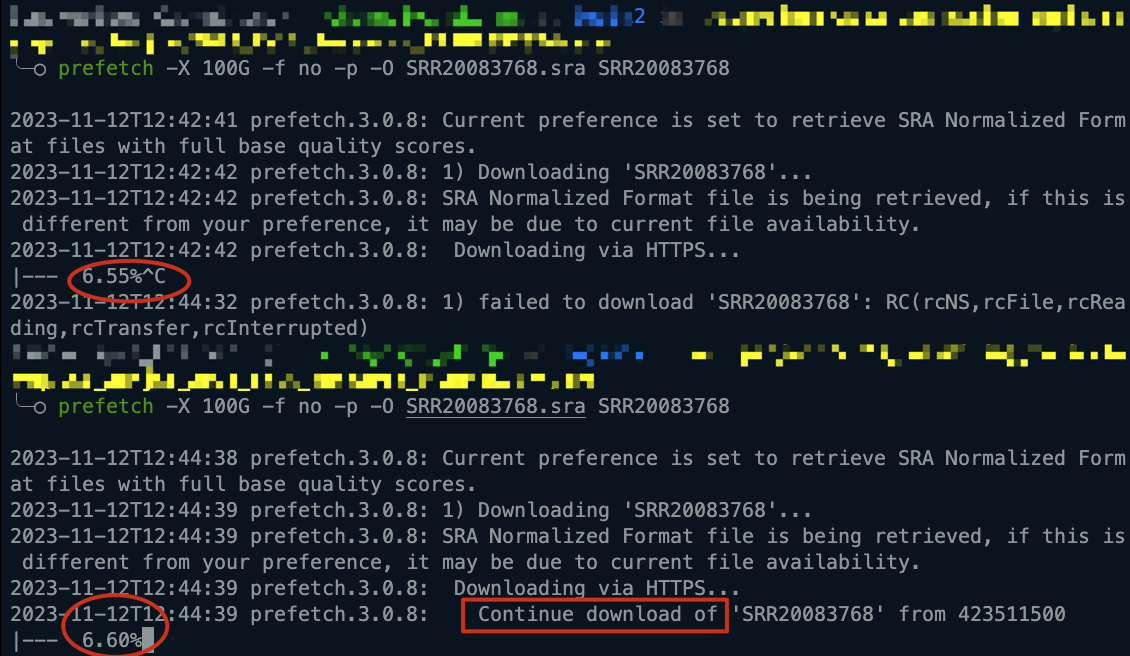
多文件下载
如果你的info.csv中有许多个文件,都需要下载,如图
 则下载示例如下
则下载示例如下
# zsh for i in `cat ../info.csv | awk -F',' '{print $1}' | sed 1d` prefetch -X 100G -f no -p -o $i.sra $i # or # bash for i in `cat ../info.csv | awk -F',' '{print $1}' | sed 1d`; do prefetch -X 100G -f no -p -o $i.sra $i done
格式转换:sra to fastq
fastq-dump, fasterq-dump和 parallel-fastq-dump用哪个?
性能测试可见:
https://zhuanlan.zhihu.com/p/591140275
而我的答案是,用fasterq-dump,理由如下,fastq-dump是初代版本,sra-tools官方工具包自带,被用了很多年,但是没有多线程,性能非常差(耗时1小时和10小时的区别);fasterq-dump同样是sra-tools官方工具包自带,近些年新加入的命令,加入了多线程,提升了很多倍性能;parallel-fastq-dump性能虽然是最强的,但是并非官方出品,兼容性和可靠性有待考证,且fasterq-dump不比它慢太多,为了流程固定,稳定,可靠,选择fasterq-dump是毋庸置疑的。
当使用conda安装完sra-tools的时候,就可以使用fasterq-dump这个命令了
fasterq-dump -h
fasterq-dump -h Usage: fasterq-dump <path> [options] fasterq-dump <accession> [options] Options: ...... fasterq-dump : 3.0.8
用例
split如何选择看这个即可
https://zhuanlan.zhihu.com/p/591140275
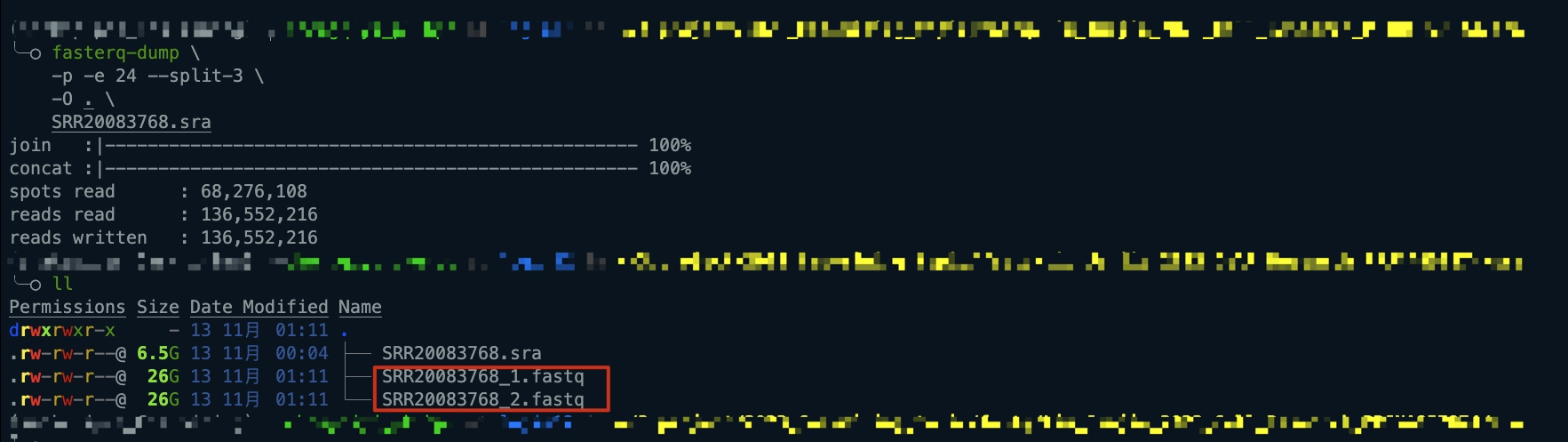
fasterq-dump \ -p -e 24 --split-3 \ -O . \ SRR20083768.sra
pigz -p 24 *.fastq
文件目录
╰─○ tree .
.
├── info.csv
└── sra
├── SRR20083768_1.fastq.gz
├── SRR20083768_2.fastq.gz
└── SRR20083768.sra
1 directory, 4 files